Retevmo was designed to target RET, the primary driver of certain RET-driven cancers1,2
Much like what has been achieved for patients with EGFR, ALK, ROS1, NTRK, and BRAF alterations, Retevmo expands treatment options for patients with certain RET-driven cancers1-4
RET alterations are a primary driver of tumor growth in the following tumor types2:
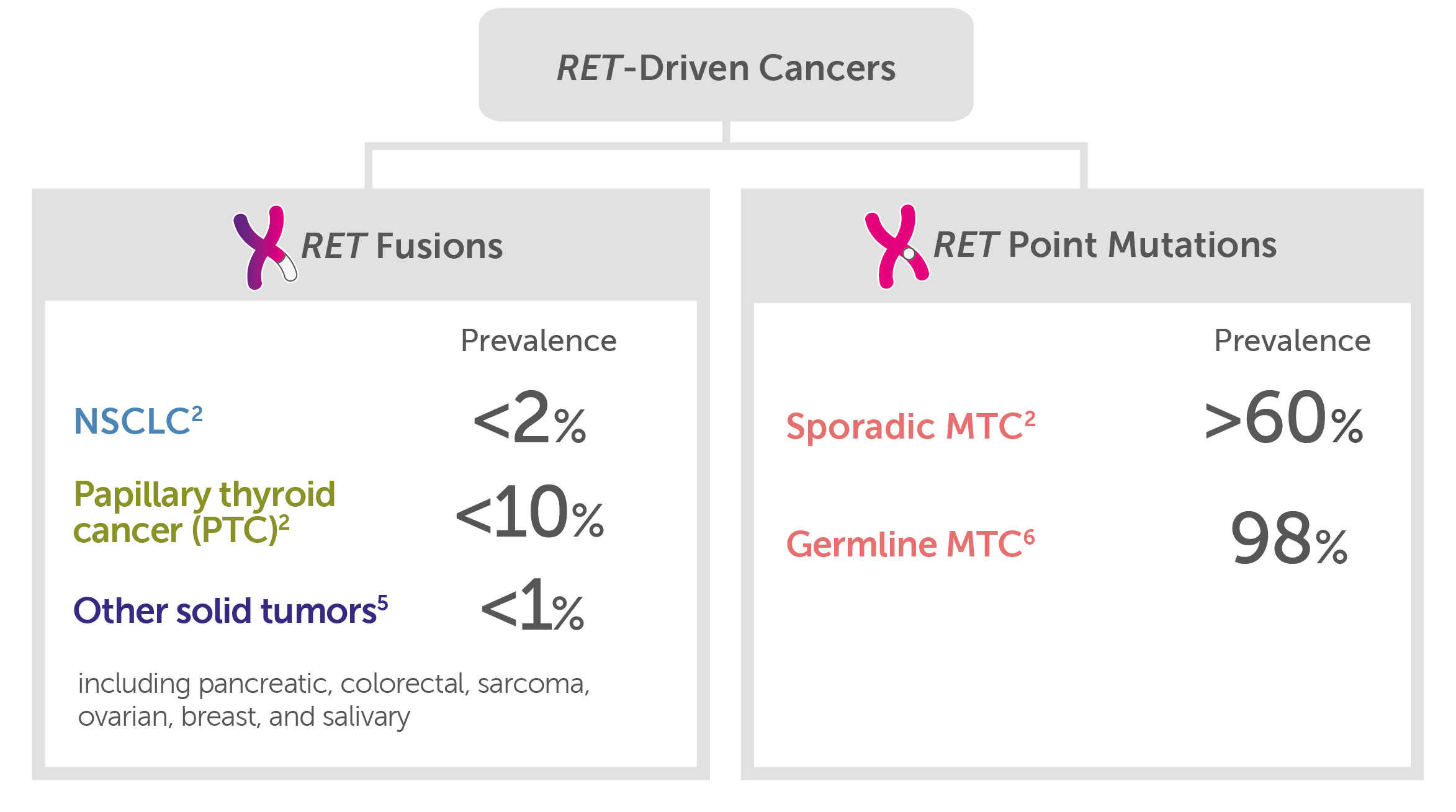
In RET-driven cancers, the prevalence of RET fusions is <2% in NSCLC, <10% in papillary thyroid cancer, and <1% of other solid tumors (including pancreatic, colorectal, sarcoma, ovarian, breast, and salivary). The prevalence of RET point mutations in medullary thyroid cancer (MTC) is >60% in sporadic MTC and 98% in germline MTC.
- RET point mutations may be present in other tumors but are believed to only drive medullary thyroid cancer (MTC)7-9
Retevmo may affect both healthy cells and tumor cells, which can result in side effects, some of which can be serious.1
RET driver alterations are predominantly mutually exclusive from other oncogenic drivers2
Most actionable genomic alterations are uncommon, highlighting the need for broad molecular profiling10,11
Select Important Safety Information
Hepatotoxicity: Serious hepatic adverse reactions occurred in 3% of patients treated with Retevmo. Increased aspartate aminotransferase (AST) occurred in 59% of patients, including Grade 3 or 4 events in 11% and increased alanine aminotransferase (ALT) occurred in 55% of patients, including Grade 3 or 4 events in 12%. Monitor ALT and AST prior to initiating Retevmo, every 2 weeks during the first 3 months, then monthly thereafter and as clinically indicated. Withhold, reduce dose, or permanently discontinue Retevmo based on the severity.
Retevmo demonstrated highly selective and potent RET inhibition1,12,13
Retevmo inhibited wild-type RET and multiple mutated RET isoforms, and was 250 times more selective for RET than 98% of ~300 kinases tested in preclinical studies1,14
Retevmo also inhibited certain isoforms of VEGFR and FGFR at higher concentrations that were still clinically achievable. In comparison, Retevmo inhibited RET at ~60-fold lower concentrations than FGFR1 and 2 and at ~8-fold lower concentrations than VEGFR3 in preclinical studies.1
Therapeutic Kinase Activity by Selectivity1,15,16

Illustration reproduced courtesy of Cell Signaling Technology, Inc. (www.cellsignal.com).
The above depicts the human kinome, which is a map of the 518 protein kinases that compose 1.7% of all human genes. The circles indicate the positions of specific kinase targets.17,18
Retevmo demonstrated potency based on phase I dose escalation data showing >90% inhibition of RET wild type and RET M918T at recommended dose.13
Retevmo was designed to selectively target an anticipated acquired resistance mechanism (V804M gatekeeper).1,2,12
Retevmo may affect both healthy cells and tumor cells, which can result in side effects, some of which can be serious.1
Retevmo crossed the blood-brain barrier and showed anti-tumor activity in mice intracranially implanted with a patient-derived RET fusion-positive tumor.1,12
ALK=anaplastic lymphoma kinase; BRAF=v-raf murine sarcoma viral oncogene homolog B; EGFR=epidermal growth factor receptor; FGFR=fibroblast growth factor receptor; MTC=medullary thyroid cancer; NSCLC=non-small cell lung cancer; NTRK=neurotrophic receptor tyrosine kinase; PTC=papillary thyroid cancer; RET=rearranged during transfection; ROS1=reactive oxygen species 1; VEGFR=vascular endothelial growth factor receptor.
References: 1. Retevmo (selpercatinib). Prescribing Information. Lilly USA, LLC. 2. Drilon A, Hu ZI, Lai GGY, et al. Targeting RET-driven cancers: lessons from evolving preclinical and clinical landscapes. Nat Rev Clin Oncol. 2018;15(3):151-167. 3. Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open. 2016;1(2):e000023. doi:10.1136/esmoopen-2015-000023. 4. Ferrara R, Auger N, Auclin E, et al. Clinical and translational implications of RET rearrangements in non-small cell lung cancer. J Thorac Oncol. 2018;13(1):27-45. 5. Yang SR, Aypar U, Rosen EY, et al. A performance comparison of commonly used assays to detect RET fusions. Clin Cancer Res. 2021;27(5):1316-1328. 6. Elisei R, Tacito A, Ramone T, et al. Twenty-five years experience on RET genetic screening on hereditary MTC: an update on the prevalence of germline RET mutations. Genes (Basel). 2019;10(9). doi:10.3390/genes10090698. 7. Mulligan LM. GDNF and the RET receptor in cancer: new insights and therapeutic potential. Front Physiol. 2019;9:1873. 8. Mulligan LM. RET revisited: expanding the oncogenic portfolio. Nat Rev Cancer 2014;14(3):173-186. 9. Kato S, Subbiah V, Marchlik E, et al. RET aberrations in diverse cancers: next-generation sequencing of 4,871 patients. Clin Cancer Res. 2017;23(8):1988-1997. 10. Hirsch FR, Suda K, Wiens J, et al. New and emerging targeted treatments in advanced non-small-cell lung cancer. Lancet. 2016;388(10048):1012-1024. 11. Suh JH, Johnson A, Albacker L, et al. Comprehensive genomic profiling facilitates implementation of the National Comprehensive Cancer Network Guidelines for lung cancer biomarker testing and identifies patients who may benefit from enrollment in mechanism-driven clinical trials. Oncologist. 2016;21(6):684-691. 12. Subbiah V, Velcheti V, Tuch BB, et al. Selective RET kinase inhibition for patients with RET-altered cancers. Ann Oncol. 2018;29(8):1869-1876. 13. Drilon A, Subbiah V, Oxnard G, et al. LIBRETTO-001: A phase 1 study of LOXO-292, a potent and highly selective RET inhibitor, in patients with RET-altered cancers. Oral presentation at: American Society of Clinical Oncology Annual Meeting. June 1–5, 2018; Chicago, IL. 14. Data on File, Lilly USA, LLC, DOF-SE-US-0004. 15. DuBois SG, Albert CM, Mascarenhas L, et al. A phase 1 study of LOXO-292, a highly selective RET inhibitor, in pediatric patients with RET-altered cancers. Poster presented at: 2019 ASCO Annual Meeting; June 1, 2019; Chicago, Illinois. Abstract TPS10066. 16. Uitdehaag JCM, Verkaar F, Alwan H, et al. A guide to picking the most selective kinase inhibitor tool compounds for pharmacological validation of drug targets. Br J Pharmacol. 2012;166(3):858-876. 17. Manning G, Whyte DB, Martinez R, et al. The protein kinase complement of the human genome. Science. 2002;298(5600):1912-1934. 18. Chartier M, Chénard T, Barker J, et al. Kinome Render: a stand-alone and web-accessible tool to annotate the human protein kinome tree. PeerJ. 2013;1:e126.
INDICATIONS
Retevmo is a kinase inhibitor indicated for the treatment of:
- adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with a rearranged during transfection (RET) gene fusion, as detected by an FDA-approved test
- adult and pediatric patients 12 years of age and older with advanced or metastatic medullary thyroid cancer (MTC) with a RET mutation, as detected by an FDA-approved test, who require systemic therapy*
- adult and pediatric patients 12 years of age and older with advanced or metastatic thyroid cancer with a RET gene fusion, as detected by an FDA-approved test, who require systemic therapy and who are radioactive iodine-refractory (if radioactive iodine is appropriate)*
- adult patients with locally advanced or metastatic solid tumors with a RET gene fusion that have progressed on or following prior systemic treatment or who have no satisfactory alternative treatment options*
*These indications are approved under accelerated approval based on overall response rate (ORR) and duration of response (DoR). Continued approval for these indications may be contingent upon verification and description of clinical benefit in confirmatory trials.